R-factor (crystallography)
In crystallography, the R-factor (sometimes called residual factor or reliability factor or the R-value or RWork) is a measure of the agreement between the crystallographic model and the experimental X-ray diffraction data. In other words, it is a measure of how well the refined structure predicts the observed data.[1] It is defined by the following equation:
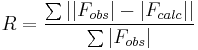
where F is the so called structure factor and the sum extends over all the reflections measured and their calculated counterparts respectively. The structure factor F is closely related to the intensity of the reflection it describes:
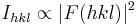
For large molecules, R-factor usually ranges between 0.6 (when comparing a random set of reflections with a given model) and 0.2 (for example for a well refined macro-molecular model at a resolution of 2.5 Ångström). Small molecules (up to 300 atoms) usually form more ordered crystals than large molecules, it is possible to attain lower R-factors. In the Cambridge Structural Database more than 95% of the 500,000+ crystals have an R-factor lower than 0.15 and 9.5% have an R-factor lower than 0.03.
Crystallographers also use the Free R-Factor (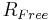 ) [2] to describe the quality of a model.
) [2] to describe the quality of a model.
The quantities 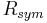 and
and 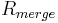 are similarly used to describe the internal agreement of measurements in a crystallographic data set.
are similarly used to describe the internal agreement of measurements in a crystallographic data set.
References
- ^ Morris AL, MacArthur MW, Hutchinson EG, Thornton JM (April 1992). "Stereochemical quality of protein structure coordinates". Proteins 12 (4): 345–64. doi:10.1002/prot.340120407. PMID 1579569.
- ^ Brunger AT (January 1992). "Free R value: a novel statistical quantity for assessing the accuracy of crystal structures". Nature 355 (6359): 472–475. Bibcode 1992Natur.355..472B. doi:10.1038/355472a0. PMID 18481394. http://www.nature.com/nature/journal/v355/n6359/abs/355472a0.html.